The basic structural units of proteins are amino acids, and are each composed of an amino group, a carboxyl group, a carbon skeleton, and a side chain of varying configurations. Phenylalanine is an essential amino acid.
Homeostasis of phenylalanine, like all amino acids, is very finely controlled, with plasma phenylalanine reflecting levels in other body compartments. Flux of phenylalanine through the plasma compartment has both input and output components. Phenylalanine input in turn may be exogenous, from the diet, or endogenous from polypeptide and free amino acid pools. The recommended dietary intake of phenylalanine varies inversely with age, reflecting increased requirements during periods of rapid growth. When nutritional intake of protein or energy is inadequate, however, catabolism results in the release of phenylalanine and other amino acids into the plasma pool (negative nitrogen balance).
Output of phenylalanine from the plasma pool can occur in one of three ways:
- incorporation into protein (which occurs in all tissues and organs);
- oxidation to tyrosine by the enzyme phenylalanine hydroxylase (only active in liver);
- conversion to minor metabolites (which takes place mainly in tissues other than liver)
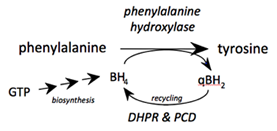
The phenylalanine hydroxylase (PAH) system is a complex reaction, being the major and rate-limiting metabolic pathway controlling the flux of phenylalanine, thereby maintaining its homeostasis. The other major component of the phenylalanine hydroxylating system is tetrahydrobiopterin (BH4), an essential cofactor which is endogenously synthesised. As a consequence of the phenylalanine hydroxylation reaction, BH4 itself is oxidised to quinonoid-dihydropterin (qBH2), and must be recycled to BH4 by the enzymes dihydropteridine reductase (DHPR) and 4 carbinolamine dehydratase for the PAH reaction to continue.
Persistent hyperphenylalaninaemia in most cases results from a primary genetic defect of PAH, with 1-2% of cases due to BH4 deficiency, because of a defect of BH4 biosynthesis or recycling (see figure). When plasma phenylalanine levels are elevated, the minor conversion pathways become important, resulting in its transamination to form phenylpyruvate (which is excreted in urine and gives rise to the term phenylketonuria), and decarboxylation to phenylethylamine.
More recently, it has been recognized that a proportion of individuals with PAH deficiency show improved phenylalanine tolerance with pharmacological doses of BH4. It is thought that this BH4 responsiveness may be because BH4 stabilises the mutant PAH protein, or because BH4 increases the expression of the PAH protein, or because the mutant PAH protein has a lower binding affinity for BH4, which is overcome by high dose BH4.
When the phenylalanine hydroxylating system is defective, tyrosine becomes an "essential" amino acid. In addition, high blood phenylalanine levels competitively inhibit the transport of amino acids across cell membranes, and cause a reduction in brain protein synthesis. This in turn may affect brain development, as well as inhibit the synthesis of the neurotransmitters dopamine, norepinephrine and serotonin. Together these biochemical aberrations may play a role in the neurological abnormalities seen in most patients with untreated hyperphenylalaninaemia.
References
Kuchel PW. Schaum's outlines: biochemistry. (3rd Ed.) New York: McGraw-Hill, 1997.
opac.library.usyd.edu.au/record=b3737352~S4
Stryer L. Biochemistry. (4th Ed.) New York: WH Freeman and Co, 1995; 647-650.
opac.library.usyd.edu.au/record=b2026517~S4
Scriver CR, Kaufman S. Hyperphenylalaninemia: Phenylalanine Hydroxylase Deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. (8th Ed.) New York: McGraw-Hill, 2001; 1667 - 1724.
opac.library.usyd.edu.au/record=b2436991~S4
E-Book 2002 at: opac.library.usyd.edu.au/record=b2773371~S4
Blau N, Thny B, Cotton RGH, Hyland K. Disorders of Tetrahydrobiopterin and Related Biogenic Amines. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. (8th Ed.) New York: McGraw-Hill, 2001; 1725 - 1776.
opac.library.usyd.edu.au/record=b2436991~S4
E-Book 2002 at: opac.library.usyd.edu.au/record=b2773371~S4
Fernandes J, Saudubray JM, Van den Berghe G, eds. Inborn Metabolic Diseases: Diagnosis and Treatment. (3rd Ed.) Berlin: Springer-Verlag 2000.
opac.library.usyd.edu.au/record=b2434909~S4
4th ED available in Badham library.